Das klinische Erscheinungsbild der HoFH kann variieren und ist essentiell für eine frühzeitige Diagnose

Sowohl Kinder als auch Erwachsene können je nach Schweregrad des Phänotyps eine Kombination der folgenden Anzeichen aufweisen
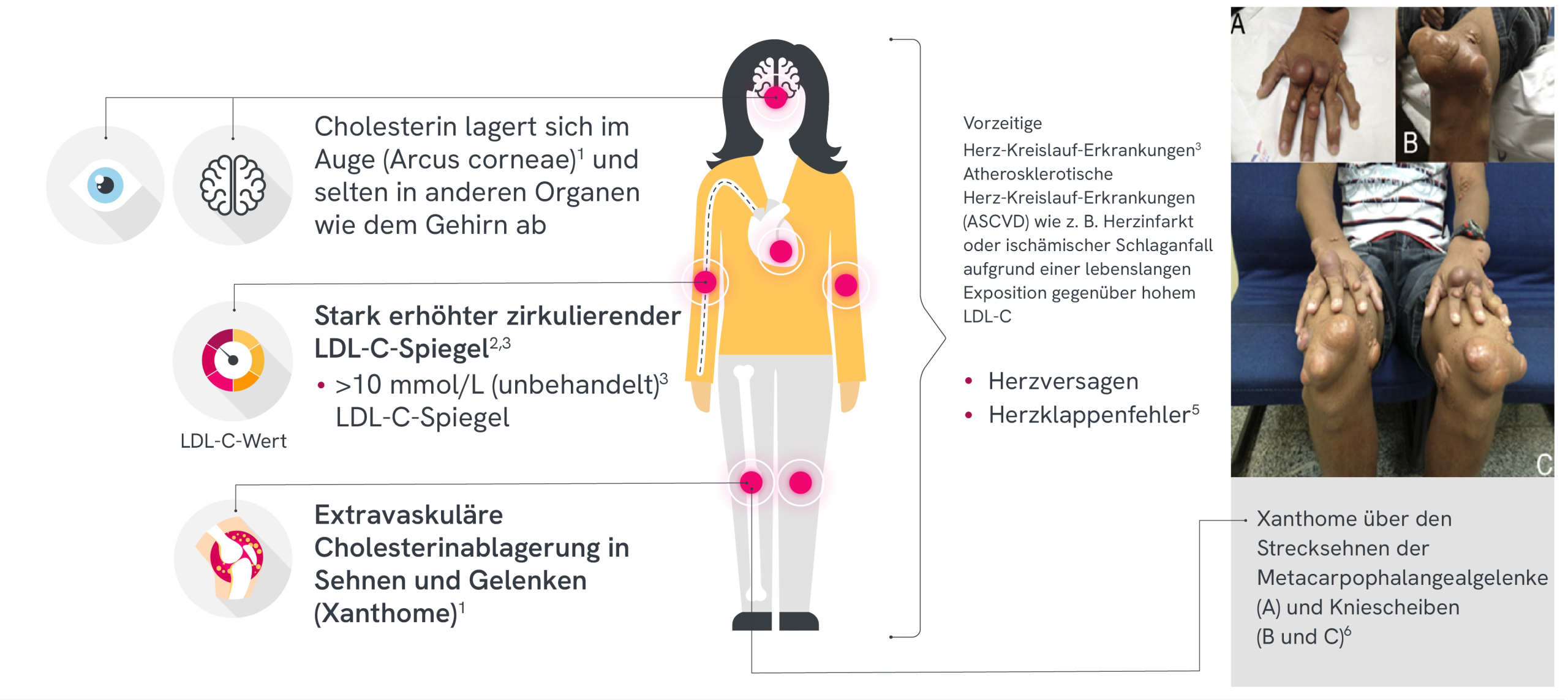
ASCVD = atherosklerotische Herz-Kreislauf-Erkrankung; CVD = Herz-Kreislauf-Erkrankung; MI = Myokardinfarkt.
ESC-/EAS-Diagnosekriterien 2023
- HoFH kann anhand klinischer und genotypischer Kriterien diagnostiziert werden, die in den EAS-Leitlinien3 erläutert werden
- Es besteht Verdacht auf HoFH, wenn der LDL-C-Spiegel im unbehandelten Zustand bei >400 mg/dl (>10 mmol/l) liegt. In diesem Fall wird eine umfassende Beurteilung der Krankengeschichte und familiären Vorbelastung und/oder ein Gentest erforderlich3
| Klinisch-diagnostische Kriterien | Genotypische diagnostische Kriterien | |
|---|---|---|
| LDL-C >10 mmol/L* | Haut- oder Sehnenxanthome vor dem 10. Lebensjahr
und/oder Unbehandelt erhöhte LDL-C-Werte, die HeFH bei beiden Elternteilen entsprechen** |
Bestätigung von zwei pathogenen Varianten im LDLR-, APOB-, PCSK9- oder LDLRAP1-Gen† |
LDLRAP1 = Low-Density-Lipoprotein-Rezeptor-Adapterprotein1
*Niedrigere LDL-C-Werte, insbesondere bei Kindern oder bei behandelten Patient:innen, können bei genetisch bestätigter HoFH beobachtet werden.
**Bei der digenischen Form können die LDL-C-Werte bei einem Elternteil normal sein und beim anderen HoFH-Werten entsprechen.
†Biallelische pathogene/wahrscheinlich pathogene Varianten auf verschiedenen Chromosomen an den Genen LDLR, APOB, PCSK9 oder LDLRAP1 oder >2 solcher Varianten an verschiedenen loci
Gentests können die Diagnose zwar unterstützen, doch HoFH kann auch bei Patient:innen ohne bekannte FH-bedingte Mutationen auftreten7
Gentests sind nützlich für die Risikostratifizierung und können bei Patient:innen mit LDLR-Mangel ein schlechtes Ansprechen auf eine medikamentöse Behandlung prognostizieren1
Bedeutung einer frühzeitigen und genauen Diagnose
HoFH wird in der Allgemeinbevölkerung unterdiagnostiziert und nicht ausreichend behandelt8,9
- Mangelnde Bekanntheit von HoFH bei Ärztinnen und Ärzten10
- Viele Fälle werden nicht im Kindesalter diagnostiziert, wenn die Entwicklung einer Atherosklerose noch vermeidbar wäre11
- Die Erkrankung wird bei einem erheblichen Anteil der Patient:innen nach der Erstvorstellung falsch diagnostiziert, wobei HoFH oft mit HeFH verwechselt wird3,12
Das Ausmass und die Dauer des erhöhten Gesamtcholesterinspiegels, der bei Patient:innen mit HoFH zu 90 % aus LDL-C besteht, ist im Allgemeinen proportional zum Schweregrad der Atherosklerose und der allgemeinen und kardiovaskulären Sterblichkeit, was die Notwendigkeit einer frühen Diagnose und Einleitung der Behandlung unterstreicht.13
Erhöhtes Risiko für allgemeine Sterblichkeit bei höherem Cholesterinspiegel (in Behandlung)13*
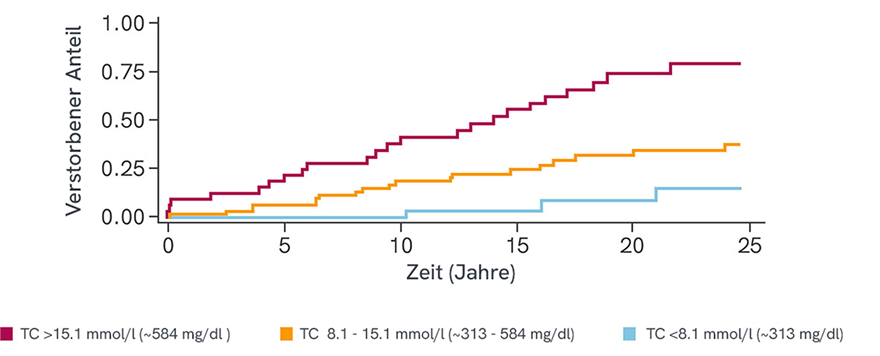
TC = Gesamtcholesterin
*Retrospektive Erhebung der Lipidwerte und klinischen Ergebnisse von 133 HoFH-Patient:innen, die von 1990 bis 2014 in Südafrika und Grossbritannien mit einer Kombination lipidsenkender Massnahmen behandelt wurden
Referenzen
1. France M et al. Atherosclerosis. 2016;255:128–139; 2. Brunham L, et al. 3. Cuchel M et al. Eur Heart J. 2023;00:1–15; 4. Santos RD et al. Lancet Diabetes Endocrinol. 2016;4:850–861; 5. Fahed AC et al. Cholesterol. 2017;2017:3685265; 6. Rocha VZ et al. J Am Coll Cardiol. 2013;61:2193; 7. Ito MK, Watts GF. Drugs. 2015;75:1715–1724; 8. Nordestgaard BG et al. Eur Heart J. 2013;34:3478–3490; 9. Baum SJ et al. J Clin Lipidol. 2014:542–549; 10. Bouhairie VE, Goldberg AC. Cardiol Clin. 2015;33:169–179; 11. Alonso R et al. J Clin Lipidol. 2016;10:953–961; 12. Hemphill L et al. J Gen Intern Med. 2020 35:2225-7; 13. Thompson GR et al. Eur Heart J. 2018;39:1162–1168. 2018;72:662-680; 11. Baum SJ et al. J Clin Lipidol. 2014:542–549; 12. Bouhairie VE, Goldberg AC. Cardiol Clin. 2015;33:169–179; 13. Alonso R et al. J Clin Lipidol. 2016;10:953–961; 14. Hemphill L et al. J Gen Intern Med. 2020 [Epub ahead of print]; 15. Thompson GR et al. Eur Heart J. 2018;39:1162–1168.
Alle Referenzen werden von Ultragenyx auf Anfrage zur Verfügung gestellt.